| Afghan Energy, Chemical & Mining Industries resource for Renewable Energies, Irrigation & Sustainable Industries. |
Urea productionThe commercial synthesis of urea involves the combination of ammonia and carbon dioxide at high pressure to form ammonium carbamate which is subsequently dehydrated by the application of heat to form urea and water.
� � � � � � � � � 2NH3 + CO2 is in equlibrium with �
NH2COONH4 in equlibrium with � CO(NH2)2 +
H2O Reaction 1 is fast and exothermic and essentially goes to completion under the reaction conditions used industrially. Reaction 2 is slower and endothermic and does not go to completion. The conversion (on a CO2 basis) is usually in the order of 50-80%. The conversion increases with increasing temperature and NH3/CO2 ratio and decreases with increasing H2O/CO2 ratio. The design of commercial processes has involved consideration of how to separate the urea from the other constituents, how to recover excess NH3, and decompose the carbamate for recycle. Attention was also devoted to developing materials to withstand the corrosive carbamate solution and to optimise the heat and energy balances. The simplest way to decompose the carbamate to CO2 and NH3 requires the reactor effluentto be depressurised and heated. The earliest urea plants operated on a "Once Through" principle where the off-gases were used as feedstocks for other products. Subsequently "Partial Recycle" techniques were developed to recover and recycle some of the NH3 and CO2 to the process. It was essential to recover all of the gases for recycle to the synthesis to optimise raw material utilisation and since recompression was too expensive an alternative method was developed. This involved cooling the gases and re-combining them to form carbamate liquor which was pumped back to the synthesis. A series of loops involving carbamate decomposers at progressively lower pressures and carbamate condensers were used. This was known as the "Total Recycle Process". A basic consequence of recycling the gases was that the NH3/CO2 molar ratio in the reactor increased thereby increasing the urea yield. Significant improvements were subsequently achieved by decomposing the carbamate in the reactor effluent without reducing the system pressure. This "Stripping Process" dominated synthesis technology and provided capital/energy savings. Two commercial stripping systems were developed, one using CO2 and the other using NH3 as the stripping gases. Since the base patents on stripping technology have expired, other processes have emerged which combine the best features of Total Recycle and Stripping Technologies. For convenience total recycle processes were identified as either "conventional" or "stripping" processes. The urea solution arising from the synthesis/recycle stages of the process is subsequently concentrated to a urea melt for conversion to a solid prilled or granular product. Improvements in process technology have concentrated on reducing production costs and minimising the environmental impact. These included boosting CO2 conversion efficiency, increasing heat recovery, reducing utilities consumption and recovering residual NH3 and urea from plant effluents. Simultaneously the size limitation of prills and concern about the prill tower off-gas effluentwere responsible for increased interest in melt granulation processes and prill tower emission abatement. Some or all of these improvements have been used in updating existing plants and some plants have added computerised systems for process control. New urea installations vary in size from 800 to 2000t/d and typically would be 1500t/d units. Modern processes have very similar energy requirements and nearly 100% material efficiency. There are some differences in the detail of the energy balances but they are deemed to be minor in effect. Block flow diagrams for CO2 and NH3 stripping total recycle processes are shown in Figures 1 and 2.
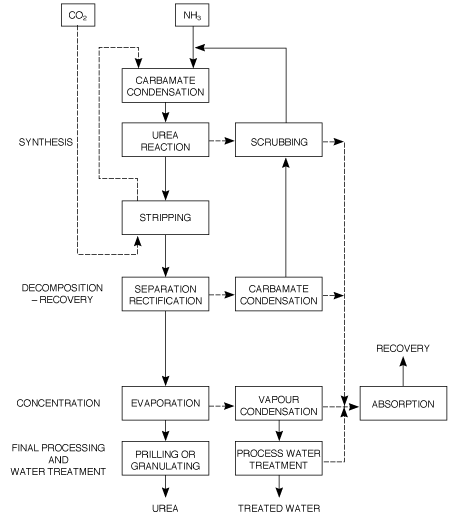
Figure 1 Block diagram of a total recycle CO2 stripping urea process.
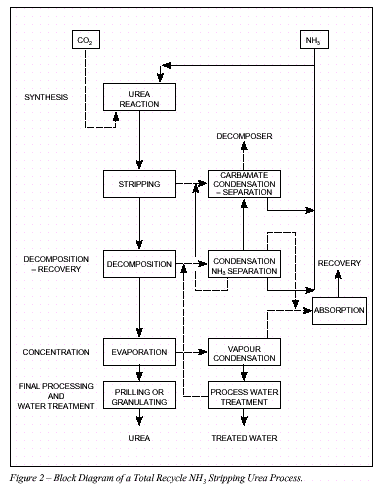
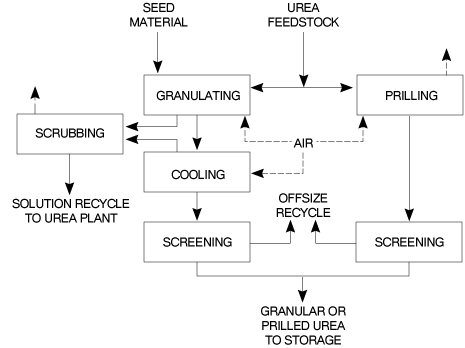
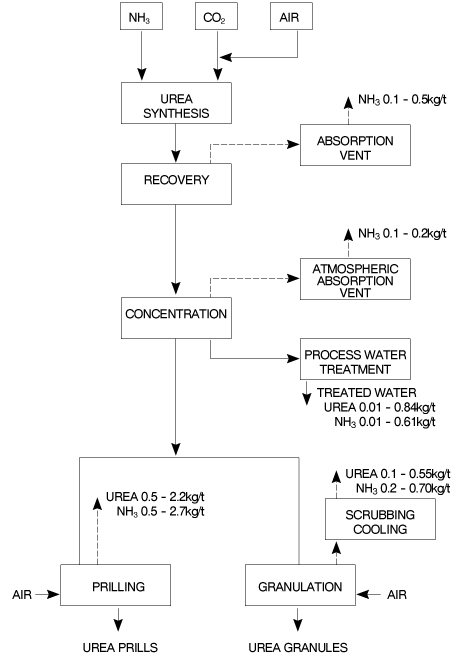
Figure 2 Block diagram of a total recycle NH3 stripping urea process 2.1 �Urea Plant Installations in Europe 24 Plants have been constructed between 1968 and 1994 in Western Europe. 7 Plants have been revamped since 1980. The total capacity of these plants is around 19,000t/d. 2.2 �Description of BAT Production Processes The process water from each process discussed in this section is purified by recovery of dissolved urea, NH3 and CO2 which are recycled to the synthesis section via a low pressure carbamate condensation system. 2.2.1 �Carbon dioxide stripping process NH3 and CO2 are converted to urea via ammonium carbamate at a pressure of approximately 140bar and a temperature of 180-185oC. The molar NH3/CO2 ratio applied in the reactor is 2.95. This results in a CO2 conversion of about 60% and an NH3 conversion of 41%. The reactor effluent, containing unconverted NH3 and CO2 is subjected to a stripping operation at essentially reactor pressure, using CO2 as stripping agent. The stripped-off NH3 and CO2 are then partially condensed and recycled to the reactor. The heat evolving from this condensation is utilised to produce 4.5bar steam some of which can be used for heating purposes in the downstream sections of the plant. Surplus 4.5bar steam is sent to the turbine of the CO2 compressor. The NH3 and CO2 in the stripper effluentare vaporised in a 4bar decomposition stage and subsequently condensed to form a carbamate solution, which is recycled to the 140bar synthesis section. Further concentration of the urea solution leaving the 4bar decomposition stage takes place in the evaporation section, where a 99.7% urea melt is produced. 2.2.2 �Ammonia stripping process NH3 and CO2 are converted to urea via ammonium carbamate at a pressure of 150bar and a temperature of 180oC. A molar ratio of 3.5 is used in the reactor giving a CO2 conversion of 65%. The reactor effluententers the stripper where a large part of the unconverted carbamate is decomposed by the stripping action of the excess NH3. Residual carbamate and CO2 are recovered downstream of the stripper in two successive stages operating at 17 and 3.5bar respectively. NH3 and CO2 vapours from the stripper top are mixed with the recovered carbamate solution from the High Pressure (HP)/Low Pressure (LP) sections, condensed in the HP carbamate condenser and fed to the reactor. The heat of condensation is used to produce LP steam. The urea solution leaving the LP decomposition stage is concentrated in the evaporation section to a urea melt. 2.2.3 �Advanced cost & energy saving (ACES) process In this process the synthesis section operates at 175bar with an NH3/CO2 molar ratio of 4 and a temperature of 185 to 190oC. The reactor effluentis stripped at essentially reactor pressure using CO2 as stripping agent. The overhead gas mixture from the stripper is fed to two carbamate condensers in parallel where the gases are condensed and recycled under gravity to the reactor along with absorbent solutions from the HP scrubber and absorber. The heat generated in the first carbamate condenser is used to generate 5bar steam and the heat formed in the second condenser is used to heat the solution leaving the stripper bottom after pressure reduction. The inerts in the synthesis section are purged to the scrubber from the reactor top for recovery and recycle of NH3 and CO2. The urea solution leaving the bottom of the stripper is further purified in HP and LP decomposers operating at approx. 17.5bar and 2.5bar respectively. The separated NH3 and CO2 are recovered to the synthesis via HP and LP absorbers. The aqueous urea solution is first concentrated to 88.7%wt in a vacuum concentrator and then to the required concentration for prilling or granulating. 2.2.4 �Isobaric double recycle (IDR) process In this process reactor pressure is about 200bar, the molar NH3/CO2 ratio is 4.5 and the reactor effluent temperature 185 to 190oC. The conversion rates to urea in the reactor are 71% for CO2 and 35% for NH3. Unconverted materials in the effluentfrom the reactor bottom are separated by heating and stripping in two consecutive decomposers operated at reactor pressure and heated by 25bar steam. Carbamate is decomposed/stripped by NH3 in the first stripper and the remaining NH3 is evolved in the second stripper using CO2 as stripping agent. The overheads from stripper 1 are fed directly to the reactor and the overheads from stripper 2 are recycled to the reactor via the carbamate condenser. Heat of condensation is recovered as 6bar steam and used downstream in the process. Most of the CO2 fed to the plant goes to the second stripper and the remainder goes directly to the reactor for fine temperature control when needed. About 40% of the NH3 goes to the first stripper and the remainder to the upper and lower sections of the reactor in two streams. Unconverted carbamate, NH3 and CO2 leaving the stripper with the urea solution are recovered/vaporised in two successive distillers operating at 20bar and 6bar respectively. The vapours are condensed and recycled to the synthesis after condensation to carbamate solution. The latent heat present in the 20bar stage off-gases is used as a heat source for the evaporation of water in the first stage evaporator. Further concentration of the urea solution leaving the LP decomposition stage is carried out in two vacuum evaporators in series, producing urea melt for prilling or granulating. 2.3 �Process Water Sources and Quantities A 1,000t/d urea plant generates on average approximately 500m3/d process water containing 6% NH3, 4% CO2 and 1.0% urea (by weight). The principal source of this water is the synthesis reaction where 0.3tonnes of water is formed per tonne of urea e.g. 2NH3 + CO2 > CO(NH2)2 + H2O The other sources of water are ejector steam, flush and seal water and steam used in the waste water treatment plant. The principal sources of urea, NH3 and CO2 in the process water are: Evaporator condensate. The NH3 and urea in the evaporator condensate are attributable to a. the presence of NH3 in the urea solution feed to the evaporator, b. the formation of biuret and the hydrolysis of urea in the evaporators, both reactions liberating NH3 2CO(NH2)2 > H2NCONHCONH2 + NH3 CO(NH2)2 + H2O > 2NH3 + CO2 c. direct carry over of urea from the evaporator separators to the condensers (physical entrainment), d. the formation of NH3 from the decomposition of urea to isocyanic acid. CO(NH2)2 > HNCO + NH3 The reverse reaction occurs on cooling the products in the condensers. Off-gases from the recovery/recirculation stage absorbed in the process water. Off-gases from the synthesis section absorbed in the process water. Flush and purge water from pumps. Liquid drains from the recovery section. � The purpose of the water treatment is to remove NH3, CO2 and urea from the process water and recycle the gases to the synthesis. This ensures raw material utilisation is optimised and effluentis minimised. 2.4 �Prilling and Granulation In urea fertilizer production operations, the final product is in either prilled or granular form. Production of either from urea melt requires the use of a large volume of cooling air which is subsequently discharged to the atmosphere. A block diagram of the prilling and granulation processes is shown in Figure 3. 2.4.1 �Prilling The concentrated (99.7%) urea melt is fed to the prilling device (e.g. rotating bucket/shower type spray head) located at the top of the prilling tower. Liquid droplets are formed which solidify and cool on free fall through the tower against a forced or natural up-draft of ambient air. The product is removed from the tower base to a conveyor belt using a rotating rake, a fluidised bed or a conical hopper. Cooling to ambient temperature and screening may be used before the product is finally transferred to storage. The design/operation of the prilling device exerts a majorinfluenced on product size. Collision of the molten droplets with the tower wall as well as inter-droplet contact causing agglomeration must be prevented. Normally mean prill diameters range from 1.6-2.0mm for prilling operations. Conditioning of the urea melt and "crystal seeding" of the melt, may be used to enhance the anti-caking and mechanical properties of the prilled product during storage/handling.
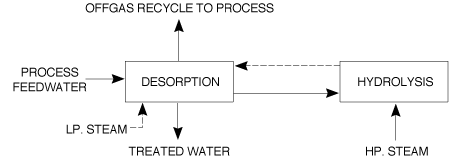
2.4.2 �Granulation Depending on the process a 95-99.7% urea feedstock is used. The lower feedstock concentration allows the second step of the evaporation process to be omitted and also simplifies the process condensate treatment step. The basic principle of the process involves the spraying of the melt onto recycled seed particles or prills circulating in the granulator. A slow increase in granule size and drying of the product takes place simultaneously. Air passing through the granulator solidifies the melt deposited on the seed material. Processes using low concentration feedstock require less cooling air since the evaporation of the additional water dissipates part of the heat which is released when the urea crystallises from liquid to solid. All the commercial processes available are characterised by product recycle, and the ratio of recycled to final product varies between 0.5 and 1.0. Prill granulation or fattening systems have a very small recycle, typically 2 to 4%. Usually the product leaving the granulator is cooled and screened prior to transfer to storage. Conditioning of the urea melt prior to spraying may also be used to enhance the storage/handling characteristics of the granular product. 2.5 Feasible and Available Emission Abatement Techniques 2.5.1 Gaseous emissions Scrubbing of off-gases with process condensate prior to venting inerts to atmosphere. Wet scrubbing of prill tower and granulation plant air to recover urea and NH3. Connection of ammonia pump safety relief valves/seals to a flare; connection of tank vents to the plant main stack or other safe location. (See 4.6.4) Dust reduction by producing granular rather than prilled product. Bag filtration of dust laden air from transfer points, screens, bagging machines, etc. coupled with a dissolving system for recycle to the process. Flash melting of solid urea over-size product for recycle to the process. Collection of solid urea spillages on a dry basis. 2.5.2 �Liquid emissions Treatment of process waste water/condensate for recovery of urea, NH3 and CO2. Improved evaporation heater/separator design to minimise urea entrainment. Provision of adequate storage capacity for plant inventory to cater for plant upset and shut-down conditions. Provision of submerged tanks to collect plant washings, etc. from drains for recycle to the waste water treatment section. Use of mechanical seals instead of gland packing for pumps. Use of closed circuit gland cooling water system for reciprocating pumps. Replacement of reciprocating machinery with centrifugal type. 2.5.3 �General Computerisation of process control to provide consistent optimum operating conditions. Implementation of regular scheduled maintenance programmes and good housekeeping practices. 2.6 �Description of Process Water BAT Treatment Systems A block diagram for a waste-water treatment plant is shown in Figure 4.
2.6.1 �Desorption hydrolysis system Heated process water is fed to the top of Desorber 1 where it is stripped of NH3 and CO2 by gas streams from Desorber 2 and the hydrolyser. The liquid leaving Desorber 1 bottom is pre-heated to 190 �C and fed at 17bar pressure to the top of the hydrolyser. 25bar steam is introduced to the bottom of the hydrolyser and under these conditions the urea is decomposed and the gases are countercurrently stripped. The vapours go to Desorber 1. The urea free liquid stream leaving the desorber is used to heat the hydrolyser feed stream and is fed after expansion to Desorber 2 where LP steam countercurrently strips the remaining NH3 and CO2 and the off-gases pass to Desorber 1. Figure 4 Block diagram for waste water treatment plant The off-gases from Desorber 1 are condensed in a cooled reflux condenser/separator. Part of the separated liquid is pumped back to Desorber 1 and the remainder is returned to the LP recirculation section of the urea plant. Residual NH3 in the separator off-gas is recovered in an atmospheric absorber and returned to the LP recirculation section also. The treated water which leaves Desorber 2 is cooled and concentrations of 5mg/l free NH3 and 1mg/l urea can be attained. 2.6.2 �Distillation-hydrolysis system Heated process water is fed to the top section of a distillation tower for NH3 and CO2 removal. The effluentliquid is pre-heated before entry to the hydrolyser where the urea is decomposed to NH3 and CO2. The hydrolyser and distillation tower vapours are mixed with off gases from the LP decomposer separator, cooled and recycled to the process. After effluenttreatment, water suitable for boiler feed is stated to be achievable. Treated water containing 5mg/l free NH3 and 1mg/l urea is expected. 2.6.3 �Stripping-hydrolysis system Heated process water containing NH3, CO2 and urea is fed to the top of a steam stripper operated at 3 bar for separation of NH3 and CO2. The water is then fed from the middle section to the hydrolyser operating at 16 bar. The gaseous overheads are then sent via the LP decomposer/absorber to the synthesis for recovery of NH3 and CO2. Free NH3 and urea concentrations of 3-5mg/l for each component are expected in the treated water. 2.6.4 �Existing emissions to water performance The actual performance of some existing plants may vary considerably from the above with values for emissions to water of 20-230mg/l (0.01-0.61kg/t of product) of NH3 and 20-320mg/l (0.01-0.84kg/t) of urea depending on the treatment system used. Figure 5 shows the emission sources from an existing plant.
Figure 5 Block diagram of emission sources and typical quantities for existing plants. 2.7 �Prill Tower Emissions The prill tower is a major source of emission in urea plants. The large volumes of discharged untreated cooling air contain particulate urea dust (1-2kg/t) as well as NH3 (0.7-1.0kg/t). 2.7.1 �Causes of dust formation
Towers with natural draft cooling are reported to have less dust emission than towers with forced/induced draft air cooling. The lower air velocity and product mass per m 3 of tower volume reduces attrition and carryover in the natural draft towers. 2.7.2 �Operation and maintenance items significantly affecting dust formation Fouling of the prilling device causing wider spread in prill granulometry. High melt feed temperature causing increased evaporation. High prill temperature at the tower base. The largest prills may not have solidified sufficiently and will fracture on impact. Dust emission is approximately proportional to prilling tower capacity. High air velocities and the air velocity distribution cause coarse dust to be entrained. Weather conditions e.g. relative humidity, temperature can affect the air quality/quantity. Unequal pressure in the prilling device causing a broad spread of prill size. 2.7.3 �Prill tower emission abatement Selection of the appropriate equipment for existing plants can be a complex issue. Dry dust collectors, irrigated electrostatic precipitators and irrigated dust scrubbers have been considered for dust abatement but few have been commercially proven. Wet scrubbers seem to be more attractive than dry dust collectors. Recovery of the NH3 from the emission (for example by aqueous scrubbing) is very � inefficient � due to the low partial pressure of the gas in the discharged air. 2.7.4 �Existing prilling plant performance Figure 5 shows the emission sources from an existing plant
2.8 �Granulator Emissions A dust emission of 5-40kg/t of final product is suggested for granulation process operations (i e ex granulator and cooler), which is is considerably higher than for prilling. 2.8.1 �Causes of dust formation The following reflects some speculations about the causes of dust formation in granulation but no quantitative data is available.
2.8.2 �Granulator emission abatement Air extracted from the plant is normally scrubbed with urea plant process condensate and the resultant urea solution is recycled for reprocessing. With standard wet scrubbers an efficiency of 98% can be achieved for dust removal. The low partial pressure of the NH3 in the discharged air results in low NH3 scrubbing/recovery efficiencies which can be increased by acidification � but the resultant solution has to be used in other plants. 2.8.3 �Existing granulation plant performance Figure 5 shows the emission sources from an existing plant.
3. Description of Storage and Transfer Equipment 3.1 �Ammonia NH3 is pumped to the urea plant at 25bar pressure and 27 �C. It is then supplied to a high pressure reciprocating pump for discharge to the urea synthesis section of the plant and the flow is regulated by a speed controller at a discharge pressure of 150-200bar depending on the process applied. The storage and transfer of ammonia are described in EFMA BAT Booklet No 1. 3.2 �Carbon Dioxide CO2 is supplied to the CO2 compressor and discharged at high pressure to the synthesis section of the urea plant. 3.3 �Formaldehyde (if used as a conditioning agent) An aqueous solution of urea-formaldehyde resin containing 50-60%wt formaldehyde and 20-25%wt urea is supplied by tanker and off-loaded to a buffer storage tank. It is injected by pump into the urea melt prior to prilling or granulation. In some modern urea granulation plants continuous urea-formaldehyde resin production units are an integral part of the granulation technology. Feedstocks are aqueous formaldehyde, molten urea and ammonia. 6. Major Hazards In urea production the following major hazards may arise:- - Equipment/piping failure due to corrosion. - Explosion hazard due to the formation of an explosive gas mixture. - Toxic hazard due to NH3 release. 6.1 �Corrosion Protection in Urea Plants Corrosion protection is achieved by the use of well proven design principles, stringent material and fabrication specifications, complemented by detailed codes of practice for operating, monitoring and inspecting equipment. The corrosiveness at a given point in the plant is determined by the temperature, the process components, the concentration of dissolved oxygen and the presence of contaminants that may accelerate corrosion. The formation on start-up and maintenance of a passive oxide layer on stainless steel surfaces is of utmost importance. Stainless steel lined carbon steel vessels are usually used in the HP synthesis section for economic reasons, including leak detection units to protect the integrity of the vessels and avoid a potentially hazardous situation. 6.2 �Explosive Gas Mixtures Explosive gas mixtures may form in the inerts scrubber, the off-gas from which consists of O2, H2, and N2 and possibly some non-condensed NH3 and CO2. Well controlled operation is a means of keeping these gas mixtures outside the explosion hazardous range. In BAT plants the hydrogen present in the CO2 feedstock is reduced by catalytic combustion to values below 10ppm, thereby minimising the risk of forming an explosive H2/O2 gas mixture in the scrubber. 6.3 �Hazard Study Urea production activities are normally integrated with an NH3 production/storage facility and are subject to the requirements of EU Directive 96/82/EC. These include the preparation of a Safety Case detailing the procedures which exist to identify and control the major hazards of loading, storage and distribution of liquid NH3. 5. Emission Monitoring 5.1 �Parameters and Frequency of Monitoring The monitoring programme adopted should include measurement of the following parameters at the suggested frequencies. A description of some of the methods available for monitoring emissions is given in Appendix 1. 5.1.1 �Emissions to air
5.1.2 �Emissions into water
5.2 �General The actual mass emission rates to air and water should be computed from the measured concentrations and flow rates. Standard methods are available for the discontinuous and continuous sampling and measurement of the emissions and should be agreed with the relevant authority. Experience has shown that sampling is the keystone of source analysis. More errors result from poor or incorrect sampling than from any other part of the measurement process. The need for continuous monitoring depends on the consistency of plant performance. 7. Occupational Health and Safety In urea plants the main chemicals to be considered for occupational health and safety purposes are ammonia, carbon dioxide, conditioning agents (e.g. Formaldehyde) and urea dust. ACGIH [2] occupational exposure limits are given in the table below. All figures are in ppmv
* �TLV-C �Threshold Limit Value Ceiling that
should not be exceeded during any part of Ammonia is a colourless gas with a characteristic pungent odour under atmospheric conditions. Carbon Dioxide is a colourless and odourless gas under atmospheric conditions. Formaldehyde may be incorporated in the final product as a conditioning agent at levels varying from 0.05% to 0.5%. Aqueous formaldehyde is injected into the urea melt and reacts to form polymeric derivatives in the matrix of the urea product. These reaction products do not have any of the hazardous characteristics of free formaldehyde gas. The healthhazards (e.g. potential animal carcinogen) associated with aqueous formaldehyde arise mainly from the gaseous formaldehyde released from the solution. Adequate ventilation must be provided to ensure that the OEL is not exceeded. General physical handling of the solution should be kept to the absolute minimum and the recommended precautions in the Safety Data Sheet adhered to. Urea dust is not regarded as hazardous. However, the general guidance (EH40 UK Health and Safety Executive) is that personal exposure to dust should be controlled to <10mg.m-3 (8 hour TWA) for inhalable dust and <5mg.m-3 (8 hour TWA) for respirable dust.
8. Summary Of BAT Emission Levels For Urea Plants The following emission levels should be achievable for new and existing plants. The levels relate to steady-state production for stand-alone units. 8.1 �Achievable Emission Levels for New Plants 8.1.1 �Emissions into air
8.1.2 �Emissions to water From Waste Water Treatment Unit
8.1.3 �Totalised emissions
8.2 �Achievable Emission Levels for Existing Plants The setting of these levels can only be dealt with on a site specific basis. The levels achievable are a function of the plant size/design/ age, the recovery systems adopted (including retrofits), the product shaping requirements (prilling or granulating) and the degree of integration with other on site processes. Some existing plants have been/can be upgraded to recover process related effluent for re-use. The main problem is the emission to air of urea dust and NH3 from the product shaping operation, particularly from prill towers. 8.2.1 �Emissions into air
8.2.2 �Emissions into water From Waste Water Treatment Unit
8.2.3 �Totalised emissions
8.3 �Solid Wastes No solid waste should arise from new or existing plants if clean or contaminated spillages are collected for re-use or sale. 8.4 �Cost of Pollution Control Measures The costs of pollution control measures in the fertilizer industry are difficult to generalise as they depend on a number of factors, such as:- - The emission target or standard to be met. - The type of process, the degree of integration with
other processes on site, production volumes, the type of raw - �Whether the plant is new so that the design can be optimised with respect to pollution abatement or whether the plant is � an existing one requiring revamping or "add-on" pollution abatement equipment. Generally, it is more economical to incorporate the pollution abatement equipment at the process design stage rather than revamping or "adding-on" equipment at a later stage. The cost of pollution control equipment for an existing plant can be 10-20% of the total cost of the plant. The operational and maintenance costs relating to environmental control can be 10-20% of the total production costs. In new plants, however, the process design would integrate environmental control with the need for high efficiency and productivity, and hence it is difficult to single out the costs of environmental control. The cost of adding-on equipment to an existing plant must be considered case by case since it is related to the size and type of plant, the type of equipment to be installed, and the pollution control requirements to be met. Hence, the costs shown below are only indicative. 8.4.1 �Feasibility of upgrading existing plants to BAT levels Treating and recovering the nutrients from prill tower effluent would require an investment of at least 6.25 million EUROs. The reduction of ammonia gas emissions requires investment in gas scrubbing and absorbing systems with additional carbamate condensing capacity for the recycle of the recovered materials to the synthesis section. The cost of these items could be at least 2.5 million EUROs. Other feasible abatement techniques include liquid spillage recovery systems, solids recovery by melting or dissolving, and additional process waste water holding capacity for up to three times the plant inventory. These areas could cost up to 2.5 million EUROs. 4. Environmental Data 4.1 �Inputs Ammonia, carbon dioxide, passivation air, conditioning agent, steam, electricity, cooling water, plant and instrument air. 4.2 �Outputs Urea, inert gases, LP steam, steam condensate, treated waste water. 4.3 �Typical Inputs for BAT Synthesis/Prilling Processes Local conditions have a major influence on optimal consumptions.
Notes 4.4 �Typical Inputs for BAT Melt Granulation Process The consumption of utilities depends to a large extent on local climatic conditions, or requirements for pollution control as well as end product temperature.
Notes 1 To re-concentrate the recovered urea solution for recycle to the process. 2 To flash-melt the over-size product for recycle to the process. 3 The use of the higher water content in the urea solution provides estimated savings of 90kg.t-1 of LP steam in the evaporation section relative to prilling. 4.5 �Production Outputs 4.5.1 �Urea Urea production in a new BAT plant is typically 1500t.d-1. 4.5.2 �Process condensate water The urea synthesis stoichiometric reaction produces process water at 0.3t.t-1 urea. Additional water sources as outlined previously may increase the final quantity to about 0.50m3.t-1 urea. The process water can be used as boiler feed water after treatment. 4.5.3 Steam condensate/turbine condensate Typically both condensates (process and steam condensates) are exported to the battery limit for polishing and re-use as boiler feed water. 4.5.4 Low pressure steam The LP steam produced in the carbamate condenser is used for heating purposes in the down stream sections of the plant. The excess may be sent to the CO2 compressor turbine or CO2 booster or exported for use in other site activities. 4.5.5 �General The actual consumption/outputs of existing plants may differ considerably from the above data. 4.6 �Production of Emissions and Waste Details of the emission sources and quantities are shown in Figure 5. 4.6.1 �Emissions into air The process steps responsible for emissions to air are:- - Urea solution formation: NH3,
CO2, inerts in scrubber vent-gas. 4.6.2 �Emissions into water The sources of NH3, CO2 and urea are as outlined in 2.3. Some older plants have been revamped to reduce emissions into air and water, and the recovered gases are recycled to the process. Newer plants have systems integrated in the original design depending on requirements. 4.6.3 �Solid waste No solid waste is produced in the urea production process. 4.6.4 �Fugitive emissions These are discontinuous releases of NH3, CO2, urea dust, formaldehyde, oil and steam. Typical sources include: storage tanks, valves including PRVs, flanges, pumps/compressor seals, sewer system vents/drains, waste water treatment units, solid urea transfer points, screens, etc. 4.7 �Environmental Hazards Associated with Emissions 4.7.1 Ammonia The molecular (undissociated) form of ammonia is highly toxic to freshwater fish and the quantity of undissociated NH3 rises markedly above pH 7.0. With fresh water, the NH3 should not rise above 25ppb to protect the most sensitive fish. Marine organisms appear far more tolerant of NH3 and it has been suggested that if the NH3 content of tidal waters is kept below 5ppm as N there is little cause for concern. However, sea water NH3 is oxidised by bacteria to nitrate and this may bring about significant lowering of dissolved O2 if it occurs on a large scale in an enclosed estuary or bay. 4.7.2 �Carbon dioxide The more CO2 in the atmosphere the more effective it is in restricting the flow of radiated energy from the earth’s surface, thereby increasing global warming. Urea production plants are extremely effective consumers of the CO2 by-product from upstream ammonia plants. 4.7.3 �Urea Urea is relatively non toxic to aquatic life. It is a natural excretory product of many marine organisms and like most nitrogenous compounds it is readily assimilated by marine phytoplankton. 4.8 �Statutory Emission Limit Values (ELVs) The statutory emission limit values (ELVs) into air normally refer to specific emissions (e.g. NH3, urea dust) from specific emission point sources (e.g. prill tower, vent, etc.). ELVs into water usually refer to the combined emissions from a site prior to discharge to the receiving water (sea, estuary or surface). No national statutory ELVs into air or water exist, for urea production units. Frequently, ELVs are negotiated between the plant/site operator and the local licensing authority. The ELVs for existing plants may reflect staged values over a defined period to enable the operator to achieve compliance. In Europe, ELVs for urea dust range from 75 to 150mg.Nm-3, and for ammonia, from 100 to 200mg.Nm-3. 4.9 �Environmental Quality Standards (EQSs) Licence conditions may also attempt to control the emissions by means of establishing EQSs which should not be exceeded in the vicinity of the plant. A time base must be stipulated whenever any limit is set (ELV or EQS) and a measuring method must be clearly defined and in most cases a frequency of monitoring for compliance must be indicated General Product Information on Urea 1. �Identification
2. �Hazards to Man and the Environment To man Urea is basically harmless when handled correctly. To the environment Urea is basically harmless when handled correctly. 3. �Physical and Chemical Properties
Appendix 1 Emission Monitoring in Urea/UAN Plants 1. �Introduction Monitoring of emissions plays an important part in environmental management. It can be beneficial in some instances to perform continuous monitoring. This can lead to rapid detection and recognition of irregular conditions and can give the operating staff the possibilty to correct and restore the optimum standard operating conditions as quickly as possible. Emission monitoring by regular spot checking in other cases will suffice to survey the status and performance of equipment and to record the emission level. In general, the frequency of monitoring depends on the type of process and the process equipment installed, the stability of the process and the reliability of the analytical method. The frequency will need to be balanced with a reasonable cost of monitoring. Particulate emissions to air will, on typical processes need to be sampled iso-kinetically. This may be done to provide a routine base-line manual check for any continuous particulate monitoring or as a routine for control purposes where continuous monitoring methods do not exist. It may be possible in some situations, to adapt the sample collection system to provide for continuous monitoring. Iso-kinetic sampling is subject to a variety of national standards and appropriate methods will generally need to be agreed with regulatory authorities. Typically they consist of combined air flow measurement and extraction sampling equipment that can be controlled to maintain the same velocity in the sampling nozzle as is present at that point in the duct. The results from checks on dry gas exhausts may then be related to on-line particulate monitoring - although this will not determine changes in aerosols. A separate analysis of the filtered exhaust gas will be necessary to measure aerosols. Wet gas systems also need to be analysed using essentially a combined iso-kinetic system with the extraction system designed to trap/separate the pollutant components for manual analysis. National standards for gas sampling systems exist and the appropriate method should be adopted. Manual methods may be necessary or accepted by the authorities in certain cases and for situations where no continuous method is available. Vent streams are not normally measured by on-line methods and when measurements are required as base-line checks, manual methods may be more appropriate. Typical methods for monitoring emissions to water rely on flow-proportioned sample collection or high frequency spot sampling together with analysis and continuous flow measurement. The use of trained staff is essential. Methods available for monitoring the emissions given in Section 8 of this Booklet are briefly described below. 2. �Emissions into Air 2.1 �NH3 and Urea Dust Commonly used methods: NH3 - Infra red spectrometry (IR). Urea dust - Transmissometer measurements or iso-kinetic sampling and gravimetric analysis of dust. 2.2 �On-Line Methods 2.2.1 �Infra red spectrometry In the simplest form of IR spectrometry the equipment consists of an optical filter, the sample cell and a detector. When the wavelength of the radiation is not selected using a prism or diffraction grating the instrument is known as a non-dispersive infra red gas analyser (NDIR), or non-dispersive ultraviolet gas analyser (NDUV), in a UV system. In a single-beam instrument a filter selects the part of the spectral range most characteristic of the substance. In a twin-beam instrument (the most commonly used instrument for on-line analysis) the radiation from the source is split and a comparison is made of the two beams after one has passed through a reference cell and the other through the sample gas. The two beams are brought together onto a half-silvered mirror or rotating chopper which alternately allows each beam to reach a detector cell which compares the heat received by capacitance or resistance measurements. The twin-beam method is preferred in an on-line system as it overcomes some of the problems associated with drift due to small changes in detector sensitivity and in the optical and spectral properties of the optical filter. However, regular zeroing and calibration are necessary to correct zero and range drift. 2.2.2 �Transmissometers Light from the source passes across the duct and is reflected by a mirror. The light beam is attenuated by the presence of particulate material in the duct and the reduction of light intensity is converted into an electrical signal which can be used to measure the concentration of particulate material in the duct. 2.2.3 �Range of Methods Available
2.3 �Manual Methods 2.3.1 �Ammonia A sample of the gas is passed through a series of absorbers containing standard sulphuric acid solution. The ammonium ions in the absorber solution may be determined by using ion chromatography, ion selective electrode or by colormetric methods. 2.3.2 �Urea dust Samples of the gas are drawn into a sampling nozzle attached directly to the inlet of a small cyclone which is inserted bodily into a gas stream at the end of the probe. The particles of grit/dust are centrifuged out of this sample and driven into a hopper. The cleaned gases are drawn from the cyclone through the probe tube, flexible hose, catchpot cooler and valve by a suction unit. The system collects substantially all dust and grit particles above 5-10 microns, when operated at sampling above 8.5Nm3.h-1 at STP. The weight of dust is gravimetrically measured in the cyclone. 3. �Emissions to Water NH3, urea or total Kjeldahl N, Biochemical Oxygen Demand (BOD), Chemical Oxygen Demand (COD), oil and metal corrosion products. 3.1 �Ammonia/Ammoniacal N The spectrophotometric method for ammonia relies on the reaction in which mono-chloramine is reacted with phenol to form an indo-phenol blue compound. This method is particularly suitable for the determination of ammonia in cooling waters derived from saline sources (dock,estuarine or sea water) and may be used in continuous flow colorimetry. Ion selective electrodes may also be used and are suitable for saline applications as well as pure water. Note that free ammonia exists in equilibrium with NH4+ as follows:- NH4+ + H2O < -> NH3 + H3O+ and that the equilibrium depends on pH. The above method determines the NH4+ ammonia. Free ammonia is particularly toxic to fish and should an incident occur, it may be more important to relate the result to free ammonia. Any suitable pH determination may be used and the free ammonia estimated as given in "Hampson B L, J Cons Int Explor, Mer, 1977,37. 11" and "Whitfield M, J Mar Biol. Ass UK, 1974,54, 562". Manual laboratory based Kjeldahl methods may be used for spot checks for the determination of organic and ammoniacal nitrogen in a mineralised sample. 3.2 �Urea (On-Line Method) The urea in the sample is chlorinated under very slightly alkaline conditions using sodium hypochlorite, sodium hypobromite and hydrochloric acid/magnesium chloride reagents. The purpose of the sodium hypochlorite is to prevent the interference of ammonia. The presence of magnesium chloride in the acid reagent is to increase the sensitivity of the method and the potassium chloride and hydrogen peroxide are to increase the rate of colour development. The method is strongly pH sensitive and so after the initial mixing of the reagent and sample the pH value of the stream is raised with borate buffer to pH 9.4. The sample is then allowed to react with an aqueous methanolic solution of phenol to form a yellow compound which is measured spectrophotometrically. 3.3 �Oil A visual inspection of the sample should be sufficient to show that no oil is present 9. Urea-Ammonium Nitrate (UAN) Production 9.1 �Overview of UAN Process Technology Ammonium nitrate (AN) and urea are used as feedstocks in the production of urea-ammonium nitrate (UAN) liquid fertilizers. Most UAN solutions typically contain 28, 30 or 32% N but other customised concentrations (including additional nutrients) are produced. Plant capacities for the production of UAN solutions range between 200 and 2000t.d-1. Most of the large scale production units are located on complexes where either urea or ammonium nitrate or both are produced. In some of the European UAN plants, ammonium nitrate is being synthesised directly from nitric acid and ammonia. In some cases carbamate solution from the urea reactor outlet is being used as feedstock for the production of UAN. In those plants the UAN technology is an integral part of the feritlizer complex. UAN from scrubbing systems, urea from sieving machines, etc. are fed to a central UAN system, where quality adjustements can be done. The addition of corrosion inhibitors or the use of corrosion resistent coatings allows carbon steel to be used for storage and transportation equipment for the solutions. West European consumption of UAN in 1998/1999 was 3.72 x 106t of solution, 41% � of which was imported. 9.1.1 �Typical UAN solution analysis N content 28-32% by weight, pH 7 to 7.5, density 1,280-1,320kg/m-3, salt-out temperature -18 to -2 �C, depending on the N content and at its lowest when the Urea N/Ammonium Nitrate N ratio is about 1:1. 9.2 �Description of Production Processes Continuous and batch type processes are used and in both processes concentrated urea and ammonium nitrate solutions are measured, mixed and then cooled. Block diagrams for UAN production are shown in Figures 6 and 7.
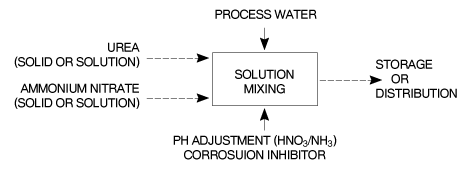
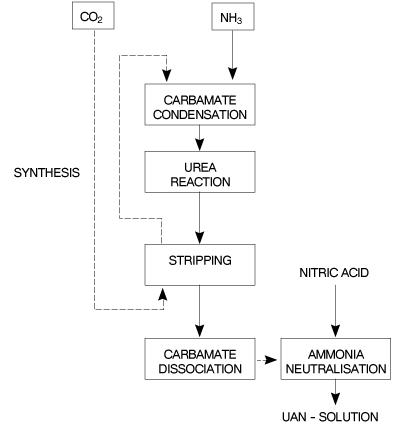
Figure 6 - Block flow diagram for UAN process.
Fgure 7 - Block diagram of a prtial recycle CO2 stripping urea process for UAN production. In the continuous process the ingredients of the UAN solution are continuously fed to and mixed in a series of appropriately sized static mixers. Raw material flow as well as finished product flow, pH and density are continuously measured and adjusted. The finished product is cooled and transferred to a storage tank for distribution. In the batch process the raw materials are sequentially fed to a mixing vessel fitted with an agitator and mounted on load-cells. The dissolving of the solid raw material(s) can be enhanced by recirculation and heat exchange as required. The pH of the UAN product is adjusted prior to the addition of the corrosion inhibitor. A partial recycle CO2 stripping urea process is also suitable for UAN solution production. Unconverted NH3 and CO2 coming from the stripped urea solution, together with the gases from the water treatment unit, are transferred for conversion into UAN solutions. 9.3 �Description of Storage and Transfer Equipment The physical form of the feedstock dictates the handling and storage system requirements. Bunded tank areas and collection pits allow any solution spillages to be collected for recycle. Air ducting and filtration helps the recovery of air-borne dust. Regulations specific to the storage and handling of solid or solutions of ammonium nitrate must be adhered to. Recommendations for the storage and transfer of ammonia and nitric acid are given in EFMA BAT Booklets Nos 1 and 2 respectively. Recommendations for the storage of solid ammonium nitrate can be found in Reference [1]. 9.4 �Environmental Data 9.4.1 �Raw material and utility inputs
9.4.2 �Typical raw materials/utilities consumption
Steam and electricity may approximate to 10-11kWh.t-1 respectively but are a function of raw material type (solid or solution) and ambient temperature. 9.4.3 �Emissions and wastes No gaseous emissions or waste arise during the non-pressure mixing of the aqueous based components. Emissions to drain are nil provided solid spillages, washings and leaks are collected in a pit or sump and recycled to the process. 9.5 �Emission Monitoring Emissions do not arise if BAT is employed. Continuous monitoring of process conditions (e.g. flow, pH, density, temperature and level) ensures optimum control and no emissions. Specific national or local requirements for monitoring may exist. 9.6 �Major Hazards The manufacture, use, storage, distribution and possession of ammonium nitrate (solid) are subject to legislation. Recommendations for its handling and storage have been issued [1]. The plant inventory of chemicals for pH adjustment (ammonia/nitric acid) will generally be too small to cause a major hazard. 9.7 �Occupational Health & Safety The materials for consideration include urea and ammonium nitrate (solids and aqueous solutions), pH adjustment chemicals (ammonia and nitric acid) and corrosion inhibitors. Full details and data for urea are given in Chapter 7 of this Booklet. Information covering ammonia, nitric acid and ammonium nitrate can be found in EFMA BAT Booklets Nos 1, 2 and 6 respectively. General product information on UAN is given in Appendix 2. 9.8 �Summary of BAT Emission Levels for UAN Solution Technologies Zero gaseous and liquid emissions are achievable for new as well as for existing UAN solution technologies.
|
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||